

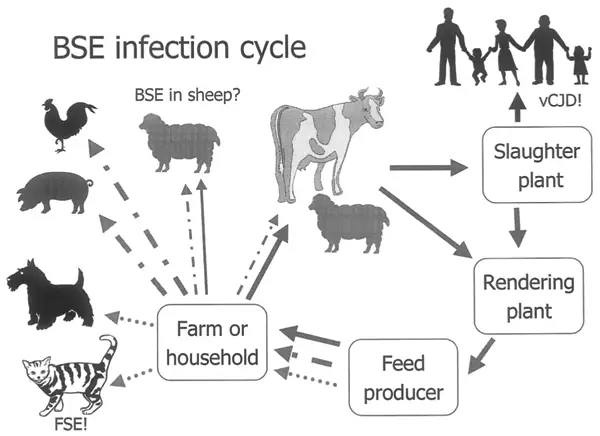
In case you want a break from SARS-CoV-2 and COVID-19, we consider something a bit different in this blog article and discuss a simpler infectious agent than viruses. Viruses are already considered non-living or not life forms, meaning they don’t fulfil the criteria for life. Yet there is something out there even simpler in its composition, but at least as feared and deadly (or perhaps more so), even compared with the most virulent and ‘dangerous’ viruses. I’m talking about prions. In between prions and viruses, there is actually another infectious agent – viroids. These are plant pathogens solely comprised on single-stranded RNA – but we won’t discuss viroids. The text commencing from the paragraph below is an extract from a report I wrote in 1997 on the molecular biology of prion diseases. Prion diseases are all neurodegenerative and infect both humans and animals alike. They can be zoonotic and are most well known as the cause of bovine spongiform encephalopathy (BSE), commonly known as mad cow disease. The foodborne connection took place in rather dramatic fashion over an approximate ten year period from the mid 1980s to the mid 1990s, with a peak during 1992 and 1993. At this time, infected beef from slaughtered cattle with BSE was identified as the cause of Variant Creutzfeldt–Jakob disease (vCJD) (Figure 1), one of the human forms of this disease. It’s reported that nearly 4½ million cattle were killed in the UK at this time (Figure 2) in an attempt to stamp out the disease by preventing infected beef from entering the food supply chain. To date, some 230 people have acquired vCJD, which has no treatment, no cure, and a 100% mortality rate. All but three reported cases were foodborne, through consumption of infected beef approximately ten years before the onset of symptoms.

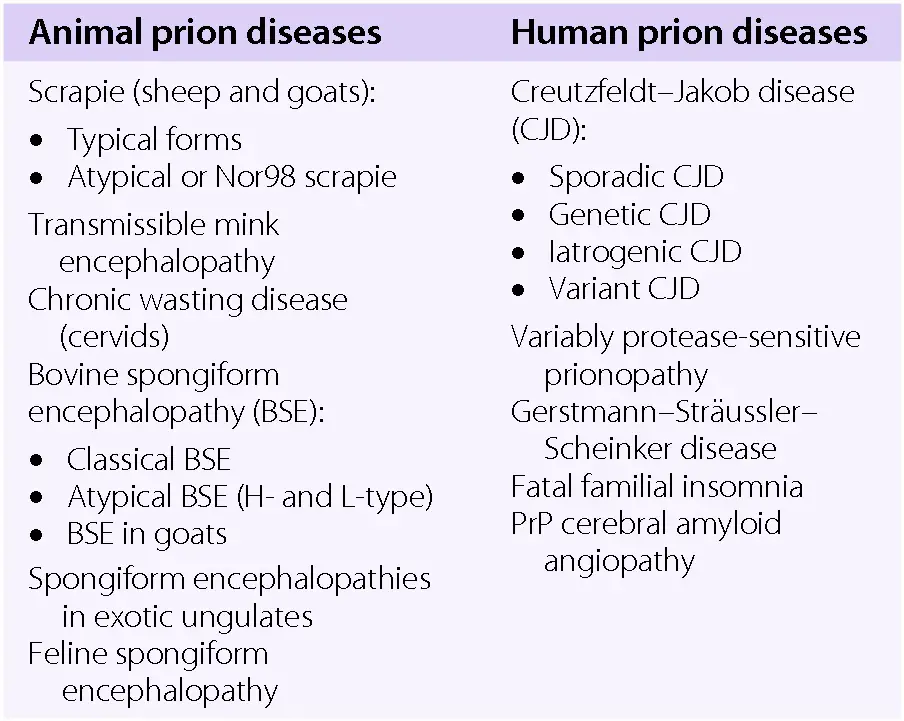
The diseases caused by prions have been known for a long time, for around 250 years in fact. Waterson and Wilkinson (1978) report that scrapie was first observed in the 1770s in certain parts of Europe, such as Great Britain, where it has been a major problem in recent history. Since then, interest has only escalated in recent years with more detailed molecular methods available for their study. We now that prions are the causal agent of ten diseases, of which four affect animals and the remaining six effect humans (Figure 3). Due to the very nature of prions, these diseases are unique (like prions themselves) in that they can be infectious and genetic. The animal diseases such as scrapie and bovine spongiform encephalopathy (BSE) have received most of the attention, although the human diseases are increasing in awareness, such as Creutzfeldt-Jakob disease (CJD) and German-Straussler-Scheinker (GSS) syndrome. Generally the animal diseases are regarded as being infectious while the human diseases are infectious or genetic, with some, like CJD being both genetic and infectious. All are characterised as being neurodegenerative.
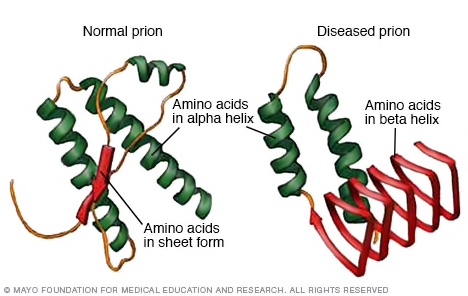
Prions, as the infectious agents that cause the above mentioned diseases lack any nucleic acid. Nucleic acid is either DNA or RNA, and is generally considered the ‘blue print’ for life, life processes or at least infectious processes. Therefore, prions are infectious proteins, or proteinaceous infectious agents, from which the name prion is derived. At the molecular level, prion diseases are centred around two forms of the prion protein, one being misfolded and infective and the other not (Figure 4). These proteins are normally produced in the body and are therefore coded for by a host gene. A proper definition of a prion would be timely, and such a definition is given by CDC (2018) who say that prions are “…abnormal, pathogenic agents that are transmissible and are able to induce abnormal folding of specific normal cellular proteins called prion proteins that are found most abundantly in the brain” – yes, a bit of a definition there to take in.
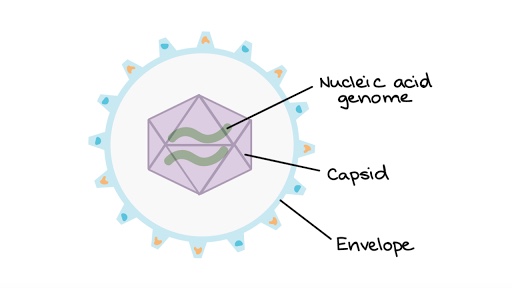
The idea of an infectious protein was first suggested by Prusiner (1982) who coined the term ‘prion’. As could be expected, this proposal was received by the scientific community with much doubt, and even in 1997, still was to some extent. However, the concept is gradually being accepted as evidence mounts. This was because every pathogen, or infectious agent, up to that time had nucleic acid, and it is the major component in the most important biological processes or replication or reproduction, basically, for continuity of the agent. Viruses were generally considered to be the simplest infectious agents (being a ‘simple’ package of nucleic acid surrounded by protein) (Figure 5), but the isolation and purification of viroids (which might be the topic of another whole blog post) and especially prions put a whole new complexion on this thinking.
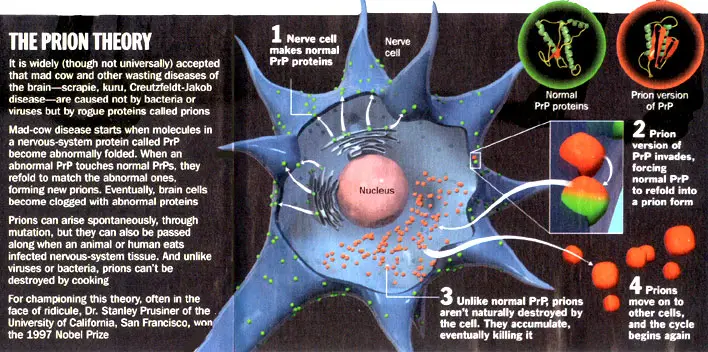
There are many fundamental differences between prions and everything else in the biological world that makes them impossible to include in any previously described category. The most significant of these is their lack of any type of nucleic acid. At first, DNA was thought to be an essential component of a pathogen. However, RNA viruses were subsequently discovered, and now we have something infectious with no nucleic acid at all. How can it survive? replicate? and be infective? All of these functions are possible as it relies on the host’s DNA. It was a very bold statement to say that an infectious agent does not contain any nucleic acid, and therefore much research was carried out to demonstrate such a theory was valid. This was done by using a variety of methods which are commonly used to degrade DNA, which were shown to not alter the infectivity of the prion protein that causes scrapie, the name of the prion disease of sheep. Many such methods were used by Prusiner (1982) in order to formulate his prion theory – for example, ultraviolet (UV) radiation and reagents like nucleases, hydroxyl amine, psoralen and zinc ions. These were not found to have any effect and neither did ionising radiation tested by Alper et al. (1978). This large amount of testing performed must surely support the theory of the absence of nucleic acid in prions. To support the theory of an infective protein, research was intensified in the early 1980s to isolate the protein. The work of Prusiner et al. (1982) and McKinley et al. (1983) identified the protein soon after the prion concept was proposed. The protease resistant core of the protein, designated PrP 27-30, was isolated from experimental hamsters, but could not be found in the control animals. Figure 6 gives a summary of what we know today or prion biology.
References
Alper, T., Haig, D.A. & Clarke, M.C. (1978). The scrapie agent: evidence against its dependence for replication on intrinsic nucleic acid. Journal of General Virology 41 : 503-516.
CDC (2018). Prion diseases. URL: https://www.cdc.gov/prions/index.html (Accessed 25 March 2020).
McKinley, M. P., Bolton, D. C., & Prusiner, S. B. (1983). A protease-resistant protein is a structural component of the scrapie prion. Cell 35 : 57-62.
Prusiner, S.B. (1982). Chemistry and biology of prions. Biochemistry 21 : 12277-12288.
Prusiner, S.B., Bolton, D.C., Groth, D.F., Bowman, K.A., Cochran, S.P. & McKinley, M.P. (1982). Further purification and characterisation of scrapie proteins. Biochemistry 21 : 6942-6950.
